Biography
I am a computational chemist who builds predictive tools and studies reaction mechanisms using DFT, molecular dynamics, QM/MM, cheminformatics, and graph neural networks. I recently (February 2026) completed my Ph.D. with Prof. Peng Liu at the University of Pittsburgh. My work focuses on three main areas:
Transition-Metal and Enzyme Catalysis
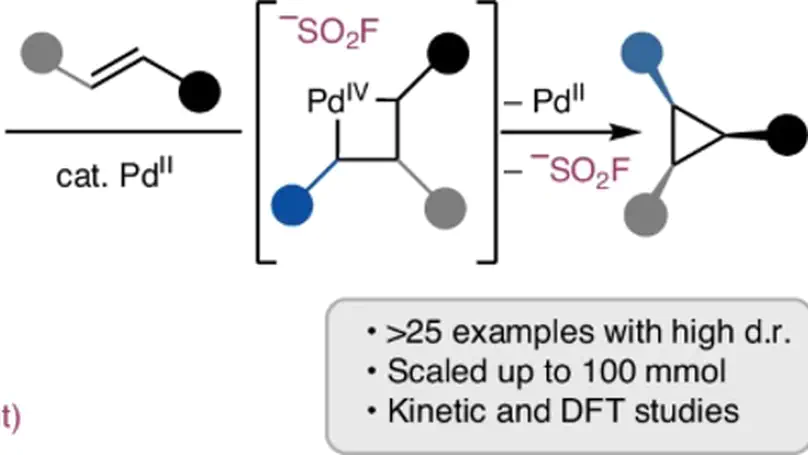
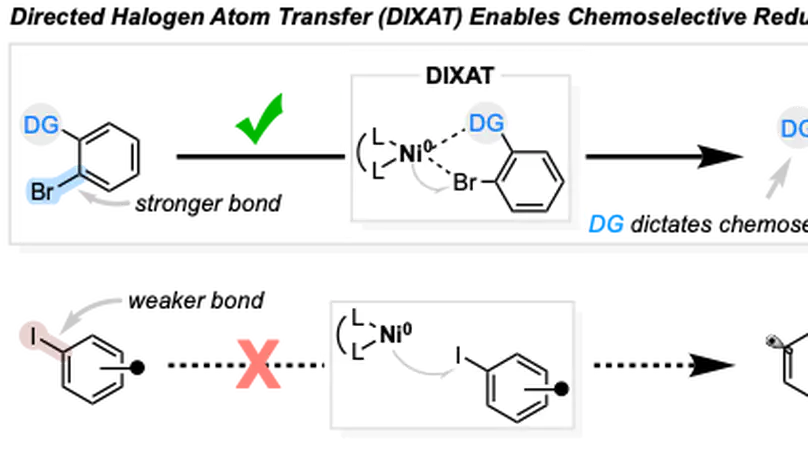
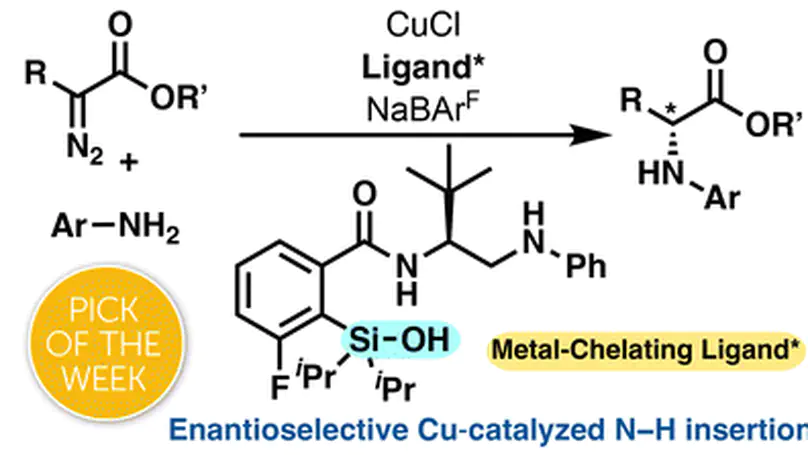
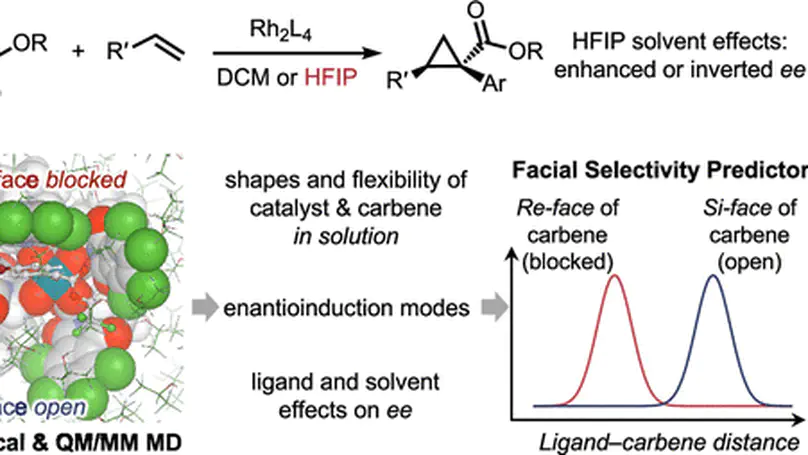
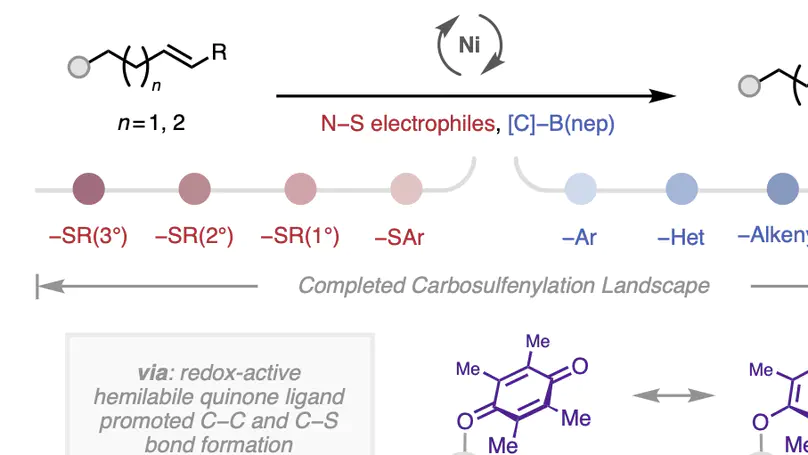
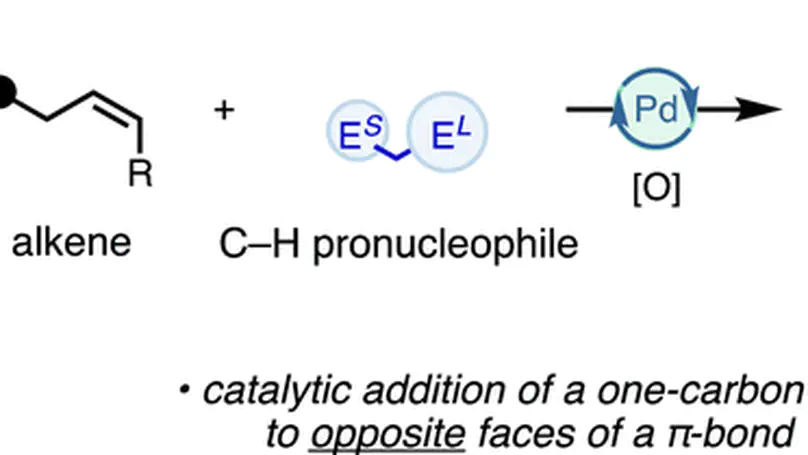
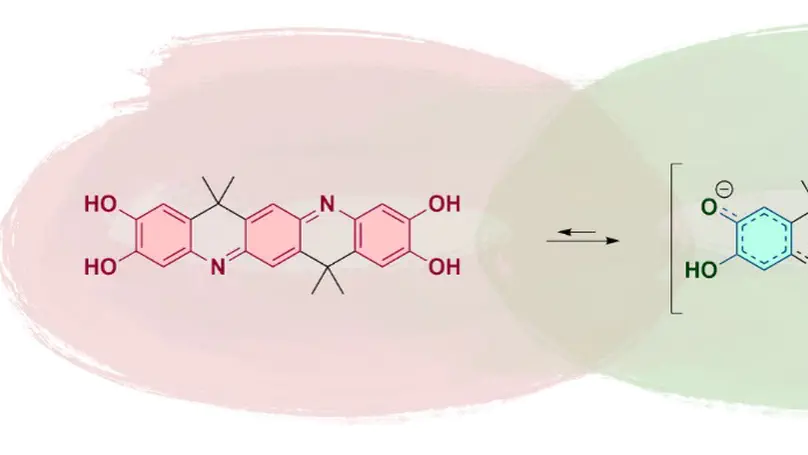
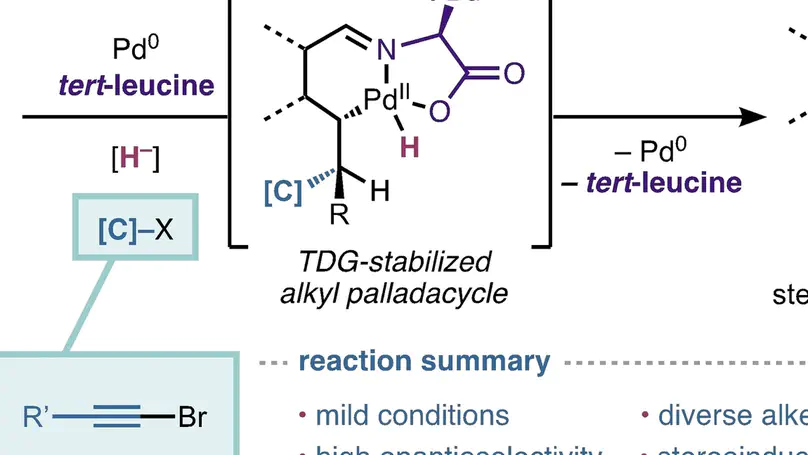
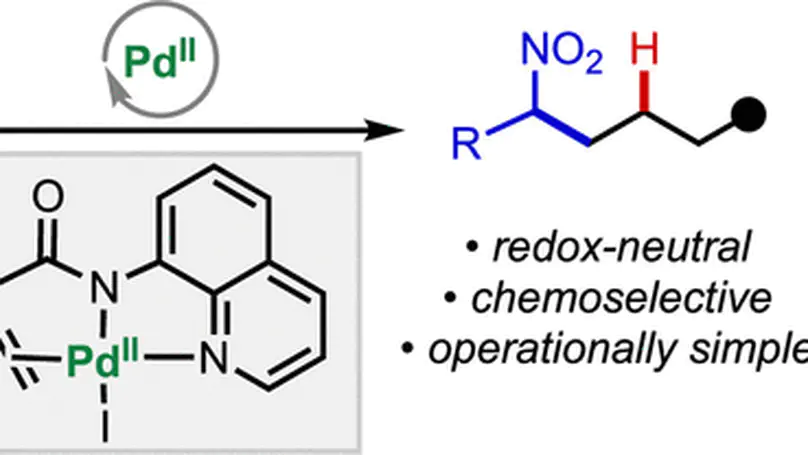
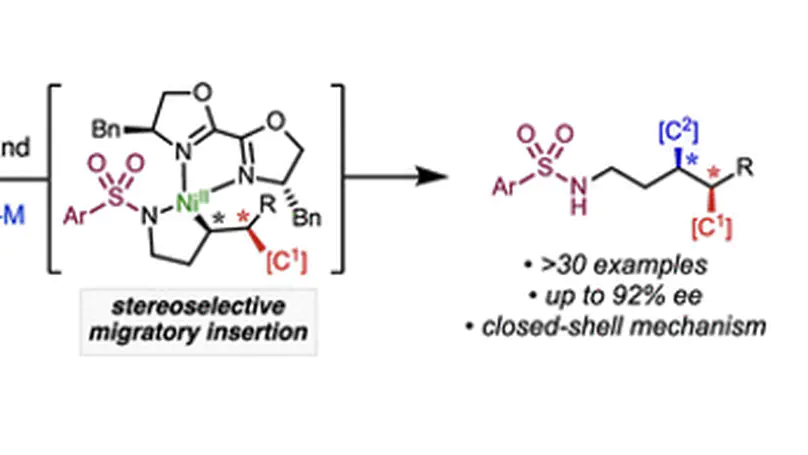
- Mechanistic Insights and Reaction Design Led computational studies on Ni/Pd/Cu/Rh-catalyzed reactions in collaboration with groups at Scripps (Keary Engle), Emory (Huw Davies), UC Davis (Annaliese Franz), UT Dallas (Vladimir Gevorgyan), and more, identifying catalyst and ligand features governing reactivity and selectivity.
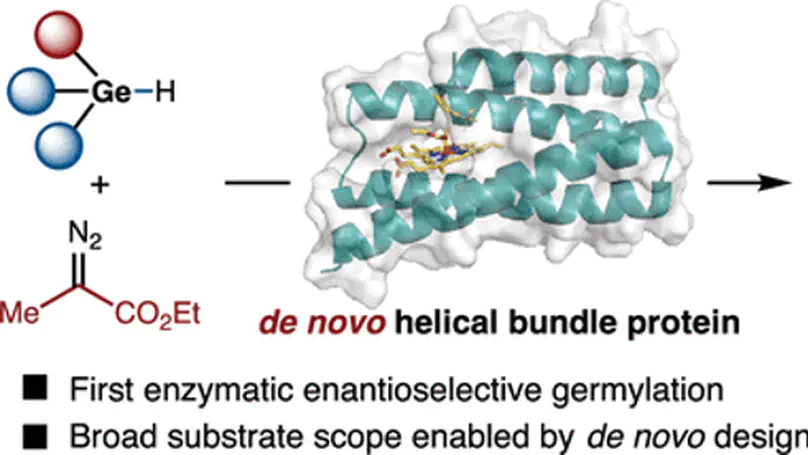
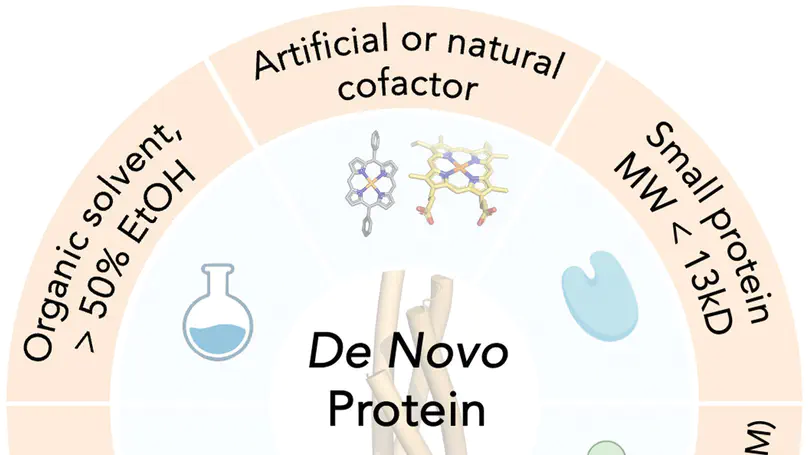
- Enzyme Design DFT/MD work with the DeGrado (UCSF) and Yang (UCSB) labs on de novo designed metalloproteins. Predicted conformer-controlled selectivity in Si-H insertion and guided directed evolution to >99:1 er for Ge-H insertion (Science 2025, JACS 2025).
Cheminformatics and Machine Learning
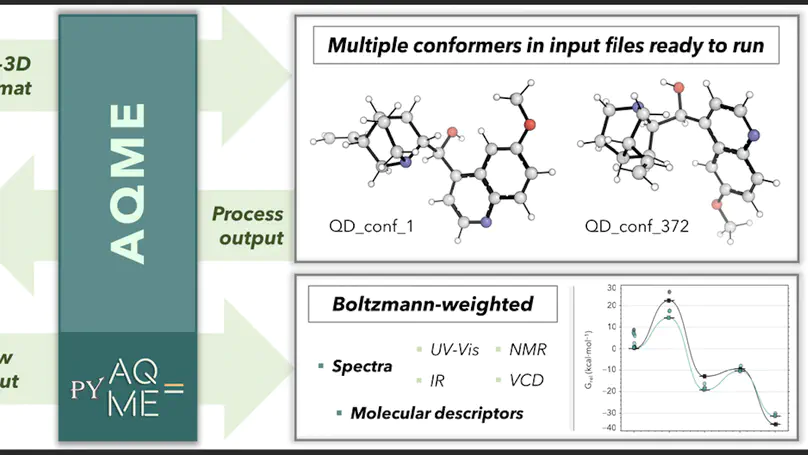
- HArD (HeteroAryl Descriptors) Built a database of >31,500 monosubstituted heteroarenes with 65 descriptors (heteroaryl Hammett parameters, aromaticity metrics, HOMO-LUMO gaps, buried volumes). Published in Sci. Data 2025. You can try the tool here: hard.pengliugroup.com
- RAPID (Radical Polarity Predictor) Trained a graph neural network on >1 million DFT calculations to predict radical polarity for any organic radical from a drawn structure. Collaboration with the Nagib and Hutchison groups. Manuscript in preparation. You can try the tool here: radicalpolarity.pengliugroup.com
High-throughput MD simulations (on-going)
- Enzyme Selectivity Prediction Exploring whether physics-based descriptors from short classical MD simulations can enhance selectivity prediction for ~900 enzyme variants, building on the MODIFY framework for ML-guided directed evolution.
Interests
- Computational Chemistry (DFT, MD, QM/MM)
- Cheminformatics & Molecular Descriptors
- Machine Learning (GNNs, Property Prediction)
- Transition Metal & Enzyme Catalysis
Education
Ph.D. in Chemistry, 2026
University of Pittsburgh
B.S. in Chemistry, 2021
Colorado State University